严禁转载!
冷冻电镜目前如日中天,今天,单粒子冷冻电镜已经不再是结构生物学领域的一种互补技术,而是逐渐成为了一种主导技术,以深刻和前所未有的方式改变结构生物学领域,促进并引领着重大新发现。今天,本文就试图从PDB数据库中一些冷冻电镜结构数据出发,来对冷冻电镜技术做一点简单的阐述。全文分6个部分来阐述:1.概述;2.冷冻电镜的发展状况;3. Cryo-EM蛋白结构分辨率的变化情况;4. 用于蛋白结构研究的冷冻电镜的主流配置;5. 冷冻电镜蛋白结构一般投稿在什么杂志上?6. 冷冻电镜解析结构的一般流程是怎样的?对样品的要求是什么?
1.概述
目前为止,能看到准确蛋白结构的,主要只有三种方法:1. X射线晶体衍射(X-ray crystallography);2. 冷冻电镜(cryo-EM);3. 核磁共振(NMR)。如下图所示,这三者的区别主要是:1. NMR可以看到蛋白的动力学,但只能看小蛋白;2. EM电镜能看很大的蛋白复合物,但是看不了小蛋白,而且(2013年以前)分辨率低,看不太清楚;3. X-ray晶体结构解析最牛,大小都能看,而且(理论上)可以看得特别清楚。X射线晶体衍射是解析蛋白质大分子和晶体结构的主要手段;Cryo-EM在降低辐射损伤,GPCR膜蛋白,难以结晶的蛋白质结构解析方面具有优势;Cryo-EM目前分辨率3埃左右,离医药研发等需求的2埃等具有一定差距;目前解析常规X射线晶体学无法解析的结构时,冷冻电镜仍是主流。冷冻电镜目前的局限在于只能做比较大的蛋白复合物,一般至少要300kDa以上才比较好做,否则蛋白太小,电镜下很容易找不到样品。另一方面,由于冷冻电镜重构的原理是对很多颗粒从不同角度拍摄最后进行叠加,所以对蛋白的均一性要求很高,如果蛋白中有一些结构可变的柔性结构也会得不到良好的结果。
顺带提一提冷冻电镜单颗粒三维重构技术原理:把一个三维的蛋白在冷冻电镜中采集到各个方向的投影图;进行二维傅里叶变换,在三维傅里叶空间重构,再进行逆傅里叶变换获得三维结构。
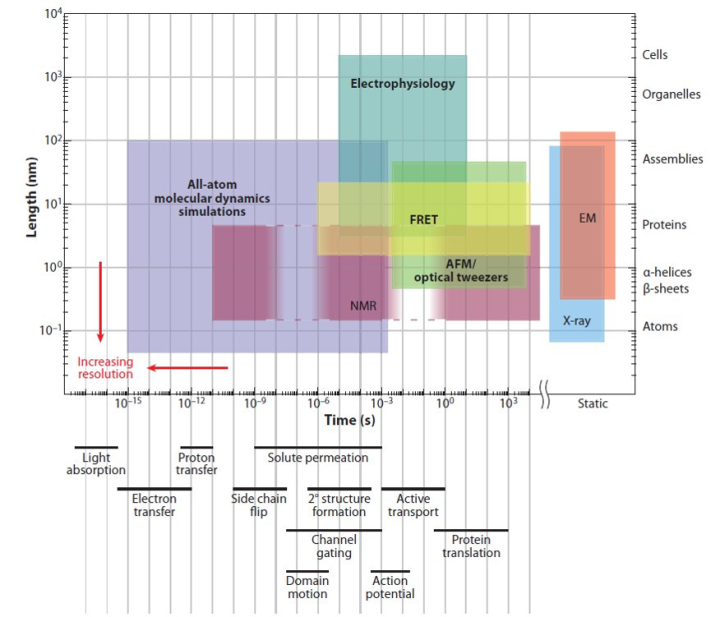
图片来自:Dror, Ron O., et al. "Biomolecular simulation: a computational microscope for molecular biology." Annual review of biophysics 41 (2012): 429-452.
2.冷冻电镜的发展状况
我们就以PDB数据库中冷冻电镜结构历年数据来做个简单的说明:
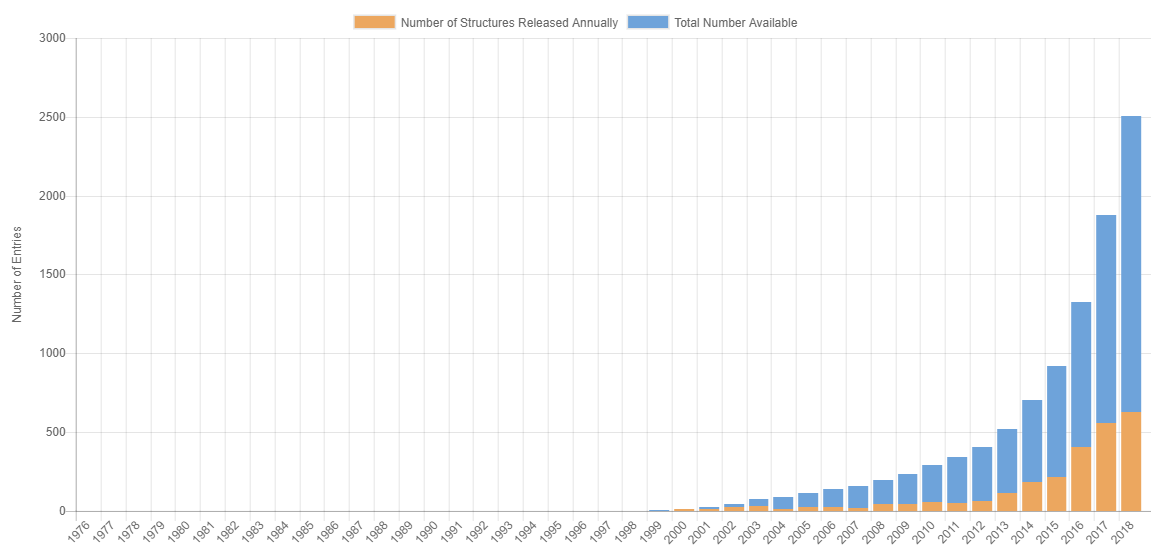
PDB数据库中冷冻电镜结构历年数据
从上图和下表可以看出,PDB数据库中Cryo-EM结构数稳步快速增加。特别是从2013年之后,Cryo-EM结构数呈现爆发式增长。这也就是为什么近几年,总是能在各大媒体上能看到冷冻电镜结构抢占头条了。
| 年份 | 电镜结构总数 | 每年新增数目 |
| 2018 | 2505 | 627 |
| 2017 | 1878 | 554 |
| 2016 | 1324 | 405 |
| 2015 | 919 | 214 |
| 2014 | 705 | 185 |
| 2013 | 520 | 115 |
| 2012 | 405 | 65 |
| 2011 | 340 | 51 |
| 2010 | 289 | 53 |
| 2009 | 236 | 40 |
| 2008 | 196 | 41 |
| 2007 | 155 | 19 |
| 2006 | 136 | 26 |
| 2005 | 110 | 23 |
| 2004 | 87 | 12 |
| 2003 | 75 | 29 |
| 2002 | 46 | 24 |
| 2001 | 22 | 9 |
| 2000 | 13 | 11 |
从上表数据上可以看到明显的一个分界点,就是2013年,这一年发生了什么事情呢?2013年,程亦凡博士与David Agard把直接电子检测相机(DDD)用于冷冻电镜单颗粒电镜图像记录。DDD直接记录电子信号,无需进行电子-光学信号转换,保持了原始信号的强度,降低了点扩散效应。同时,冷冻电镜图像能够被记录为一堆电影帧,每个电影帧都在短时间内被记录。由于速度快,也能减少了样品漂移产生的图像模糊,提高电镜图像的分辨率。用DDD相机记录的图像可以保持高频信号确定高分辨率结构,保持低频信号用于图像对准所需的对比度。因此,用DDD纪录的图像分辨率高、信噪比高、信号强、读出速度快,解决了一直限制cryo-EM发展中的两个最困难的问题,使得许多生物大分子复合物可以以原子分辨率重建3D密度图。
所以,从2013年之后,Cryo-EM在结构生物学领域的成果数强劲攀升。现在(2018年10月) 每年有将近600-700个Cryo-EM测定的蛋白结构提交到PDB数据库。
我们再看看,这些年Cryo-EM测定的蛋白结构的分辨率是一个什么变化情况。
3. Cryo-EM蛋白结构分辨率的变化情况
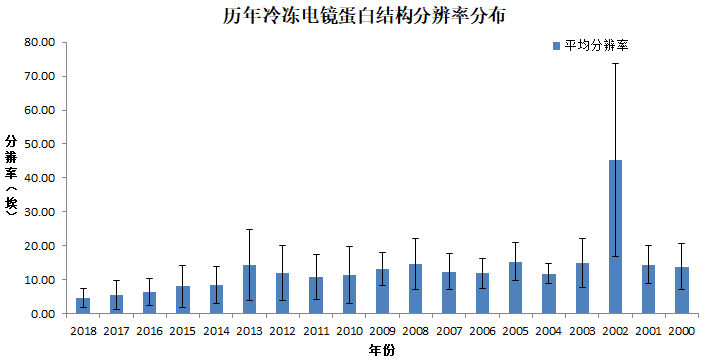
| 年份 | 平均分辨率(埃) | 标准差 |
| 2018 | 4.58 | 2.73 |
| 2017 | 5.58 | 4.29 |
| 2016 | 6.34 | 4.07 |
| 2015 | 7.99 | 6.15 |
| 2014 | 8.53 | 5.48 |
| 2013 | 14.35 | 10.43 |
很显然,从13年有了技术突破后,14年开始,Cryo-EM测定的蛋白结构的分辨率在不断的提升。其中,2018年的结构当中,分辨率的分布情况是
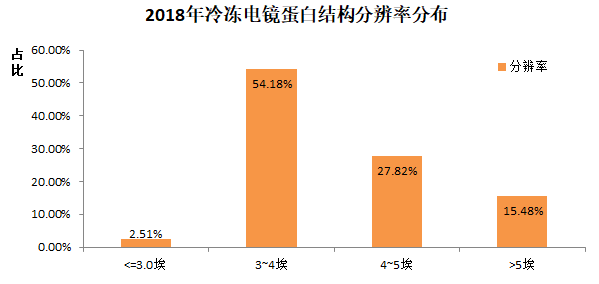
值得注意的是,目前分辨率3-4埃左右,离医药研发等需求的2埃等具有一定差距。
4. 用于蛋白结构研究的冷冻电镜都有哪些型号,主流配置是什么?

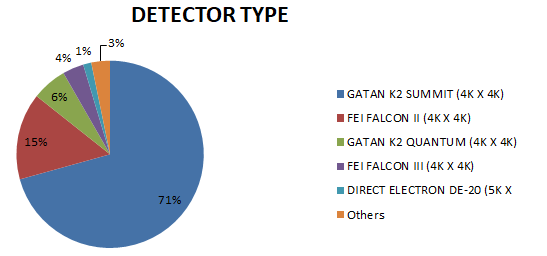
上图我们统计了2017年-2018年,PDB数据库中所有电镜结构的仪器配置。很明显,目前主流的冷冻电镜是300kV冷冻透射电子显微镜Titan Krios,再配上FEI FALCON II (4K X 4K)检测器。据了解,目前国内配置(含正在招标、筹建的)该种型号冷冻电镜的大学和科研院所有:清华大学,北京大学,浙江大学,复旦大学,南方科技大学,上海科技大学,四川大学。安装好200kV电镜的大学有南开大学,西安交通大学,兰州大学,吉林大学,中山大学,厦门大学等。
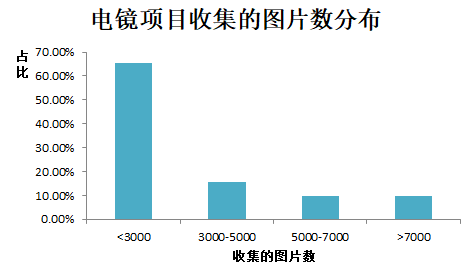
那么对于重构一个蛋白结构,我们需要收集多少张图片呢?我们也做了统计,分布如下:80%以上的项目只需要收集5000张以下的图片即可。一般来说,图片数越多,分辨率会有所提高,但是也就意味着数据收集的费用也越高。
5. 冷冻电镜蛋白结构一般投稿在什么杂志上呢?
毫不委婉的来说,作为科研人员,我们最关心的还是科研成果能发表在什么档次的杂志上。下图和下表是我们统计的2017-2018年电镜结构投稿的杂志分布情况。NATURE, PROC. NATL. ACAD. SCI., ELIFE, NAT COMMUN, NAT. STRUCT. MOL. BIOL., SCIENCE, CELL, MOL. CELL, STRUCTURE这几大杂志占据了80%的份额。(注:此数据统计的是PDB结构关联的杂志名称,有可能多个PDB ID对应一篇文章,所以可能会与实际情况有所出入。)
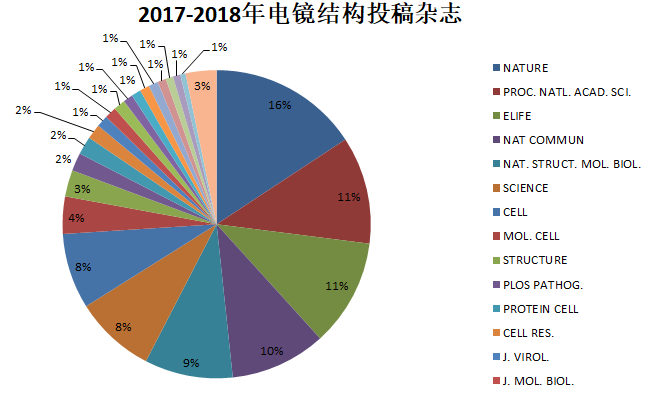
| 杂志名称 | 电镜PDB ID数 |
| NATURE | 141 |
| PROC. NATL. ACAD. SCI. | 101 |
| ELIFE | 101 |
| NAT COMMUN | 90 |
| NAT. STRUCT. MOL. BIOL. | 83 |
| SCIENCE | 76 |
| CELL | 71 |
| MOL. CELL | 35 |
| STRUCTURE | 25 |
| PLOS PATHOG. | 17 |
| PROTEIN CELL | 17 |
| CELL RES. | 14 |
| J. VIROL. | 11 |
| J. MOL. BIOL. | 11 |
| NAT MICROBIOL | 11 |
| CELL REP | 9 |
| NUCLEIC ACIDS RES. | 9 |
| J. BIOL. CHEM. | 9 |
| NEURON | 9 |
| SCI ADV | 8 |
| SCI REP | 7 |
| BIORXIV | 7 |
| EMBO J. | 5 |
| Others | 29 |
6. 冷冻电镜解析结构的一般流程是怎样的?对样品的要求是什么?难度在哪?
冷冻电镜解析蛋白结构一般流程为:
蛋白表达纯化
负染样品准备:约2小时完成
负染样品的数据收集:约8小时完成
冷冻样品的准备:约4小时完成
冷冻样品的数据收集:48-120小时完成
三维结构重建。
冷冻电镜解析蛋白结构对蛋白质的要求:
分子量:一般需要样品的分子量在200kD以上
缓冲液:缓冲液中不能含有多糖,DMSO,甘油等有机物质,这些会降低样品的衬度,难以获得高分辨的三维结构。一般而言,缓冲液为20mM Hepes,150mM NaCl。
浓度:一般而言,可溶性蛋白浓度应在1mg/ml左右,膜蛋白应保证浓度在5mg/ml左右。
体积:20ul足够(前提是需要蛋白浓度达标,做一个样品3ul左右)。
均一性:分子筛行为表现为单一的峰,均一性大于90%。
说明:冷冻电镜解析结构主要风险在:
A. 样品很不稳定,样品寄送过来的时候,已经降解或者聚集,无法进行后续处理;
B. 样品在冷冻制样的过程中,可能会被冻碎,从而无法进行后续处理;
C. 样品纯度可能很好,但是均一度很差,从而难以获得样品的高分辨结构;
D. 我们关心的区域可能在整个复合体中有很强的柔性,从而经过二维或者三维平均后,我们关心区域的分辨率会比较差甚至看不见,达不到我们的预期;
E. 一些配体,比如药物前体分子,分子量太小,不一定能在电镜的密度图中观测到;
F. 对于合适的样品,我们冷冻制样需要优化的参数有很多,包括样品浓度的优化,blot time的优化,温度的优化,grid规格(铜网或者金网)的优化等一系列条件的优化。
因此冷冻电镜制样需要丰富的经验和充足的机时支持,不同的实验者,成功率可能差别很大。
实例展示

上图这个样品似乎被DNA污染了

这个样品样品冷冻出现问题,出现冰晶

这个样品就比较理想了
Souce: 纽普生物 2018-10-29