之前我们用了AutoDock 4写了个实例教程,教程地址:http://www.novopro.cn/articles/201712271167.html
今天我们就用一个号称精度更高,对接速度更快的软件AutoDock Vina做一个案例讲解。

这图表示用AutoDock4对接的话,有50%的概率是比较准的,还有50%的概率不太准。而用AutoDock Vina对接的话,比较准(衡量的标准是RMSD<2埃)的概率提高到了78%。备注:RMSD衡量的是预测结果与真实结果中原子之间的距离偏差。
我们还是以之前教程中的3uf0为例,来看看vina的准确性如何,同时如何提高对接的准确性。
首先来看看3uf0模型中真实的结合方式是怎样的,见下图:

好了,我们开始用Vina做对接了。
0. 用到两个程序:vina.exe和vina_split.exe,其中vina.exe是用来做对接的而vina_split.exe是用来分割对接结果的。这两个程序都可以在http://vina.scripps.edu/下载得到。
1. 跟AutoDock4对接一样,在mgltools中准备好pdbqt格式的受体和配体分子,不在赘述
2. 创建一个配置文件:3uf0.conf,这个文件里面写上对接参数。详细参数如下:
receptor = r.pdbqt
ligand = nap.pdbqt
center_x = 32.961
center_y = 7.86
center_z = 25.85
size_x = 98.00
size_y = 126.00
size_z = 96.00
energy_range = 4
exhaustiveness = 9
num_modes = 9
稍微说下参数的意义:
receptor:指定受体分子的路径
ligand:配体分子的路径
center_x,center_y,center_z:搜索空间中心的坐标
size_x,size_y,size_z:指定搜索空间的大小。这里设置的大小基本就是把整个受体分子都包含了,属于blind docking。如何更准确确定结合口袋的位置,我们稍后再说。
energy_range:与最优结合模型相差的最大能量值,单位是kcal/mol。比方说,最优模型的能量计算出来是-8.5kcal/mol,那么vina也就最多计算到-4.5kcal/mol的模型就终止了,也就意味着这个值决定了生成模型的最大个数。
exhaustiveness:用来控制对接的细致程度,默认值是8. 大致与时间成正比。
num_modes:最多生成多少个模型。实际生成的模型数由num_modes和energy_range共同决定。
3. 把配体分子,受体分子,参数文件,vina程序都放在同一个目录中。比如:E:\AutoDock\3uf0

4. 命令行进入到该目录,输入:vina --config 3uf0.conf,回车

几分钟后,vina对接出了9个模型,如下:

5.我们挑选第一个模型,看看结构方式是怎样的,见下图。很显然与真实的结合方式相差甚远,可以说是完全错误。

为什么会出现这种情况,很大程度上是因为search space太大,可能需要设置更大的exhaustiveness。
如果我们大致知道binding pocket在什么位置,那准确性应该会高不少,如何大致确定binding pocket的位置呢?我们接着试验。
可以通过实验的方式,比如某个点突变对结合或者活性影响非常大,那么大概率这个残基是结合口袋的一部分。
可以通过软件预测,比如蛋白与配体(底物)结合位点预测:https://zhanglab.ccmb.med.umich.edu/COACH/
再比如Discovery studio软件(专业版的),可以很方便的根据受体分子的表面形状来预测结合口袋位置。

口袋的坐标为:
34.3356,14.9412,26.9615
我们修改下对接参数,新的参数如下:
receptor = r.pdbqt
ligand = nap.pdbqt
center_x = 34.3356
center_y = 14.9412
center_z = 26.9615
size_x = 30.0
size_y = 30.0
size_z = 30.0
energy_range = 4
exhaustiveness = 10
num_modes = 10
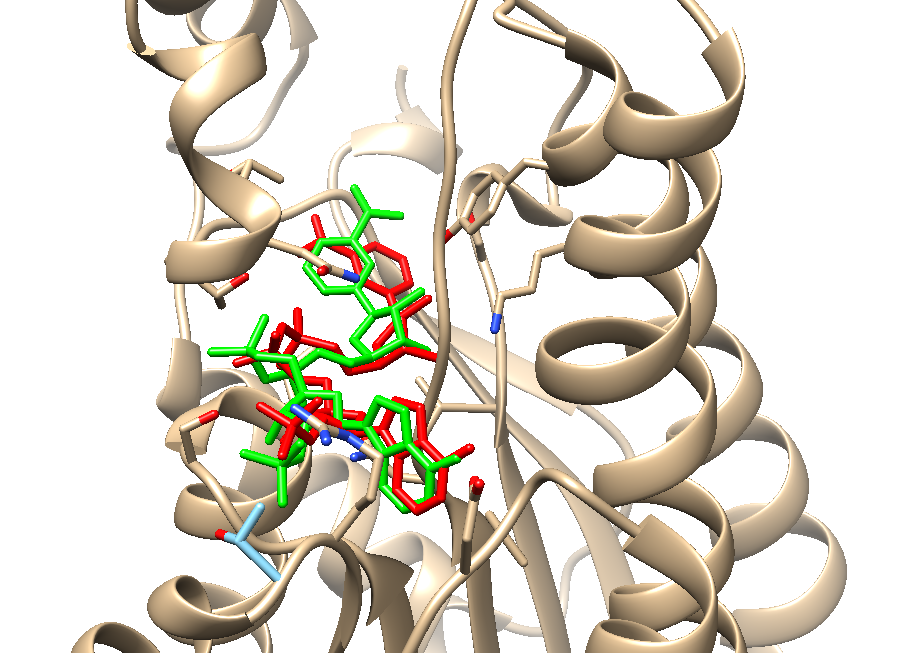
最优结果与真实模型的RMSD为1.795埃,可以说非常精准了
比对结果如下:
Souce: 纽普生物 2019-05-07